
“We are Here” - AI and Drug Discovery by Jeremy Jenkins, Novartis’ Head of US Discovery Sciences
A field note from Dr. Jeremy Jenkins's BioIT World 2026 plenary on where AI and drug discovery actually stand today. Not the hyped version, not a future projection. The current state of AI implementation, from inside Novartis.
We are Here
A few years ago the Bio-IT World plenary would go to a person debuting a new data model architecture; this year it went to Jeremy Jenkins of Novartis, who used over 45 slides to describe what works inside a global top-tier pharma. His talk was titled “Generative AI Across Drug Discovery Tasks,” but the title undersells the shift he describes in his presentation. This was a move from architecture to operation, from theory to practice.
His assessment of progress and current status was remarkable for placing where we are in context of what went before, and kept the scope of his talk to three specific areas of drug discovery and development: target identification, small molecule design, and assay development.
Laying down context
Of course there are many other steps in the ~13 year time it takes to take a drug from a disease model to final approval. One thing Dr. Jenkins pointed out, was that when machine learning was first referenced in 2004 for drug discovery, there were no subsequent references to “machine learning drugs”; he points out how foolish it is in the current euphoria over AI to refer to “AI drugs”. (Nonetheless, as an aside, there are some 15 drugs currently in Phase III clinical trials characterized as “AI-discovered”.)
He then pointed out how a determinative model differs from a generative one; the generative one abstracts underlying patterns from the input (“training”) data, to generate new data. The product is a probability distribution. He then showed how language prediction for LLMs can be transformed into “image to image translation”, where a brightfield image can predict the presence of specific proteins of interest. (He foreshadows here what he’ll cover in the third part of his presentation.) Or a transformation of “chemical space” where out of the some 1060 potential chemical compounds, a reference molecule can serve as the known, and the unknown “prior” molecule can be iteratively moved toward the reference molecule with increasing bias toward the reference (this is the second portion of his presentation).
At the bottom of this explanation was a useful footnote: “Not covered today…” (insert long list of areas where AI could be applied, including PK/PD predictions, dose selection, document generation) “…and MANY MORE”.
As an aside, personally I often encounter friends who feel strongly there is an “AI Bubble” where there are overenthusiastic investors are on one side and hyperventilating journalists and alarmed citizens on the other. I point them to insatiable demand for AI services, whether it is Anthropic reporting annual run-rate revenue of $30B or this footnote.
In genomics there are parallels where “AI Sequencing” may appear as a monolithic product category, but are actually several distinct sub-domains such as AI variant calling, AI mutation pathogenicity prediction, and AI sequencing alignment among others.
Target Identification
The first area discussed was target identification (“picking the right target molecule”), where Novartis uses four canonical approaches: tissue expression and functional genomics, human genetics, current biological knowledge (that is, published literature; Novartis uses the LLM tool Causaly which happened to be a major sponsor), and in silico perturbations of single-cell foundation models.
It is the biology where drug discovery begins. And the target identification is a very hard first step.
Interestingly, he spent no time at all on the first three (functional genomics, human genetics and biomedical literature) and went straight to perturbations of single-cell models. Perhaps because the first three areas are the traditional approaches, and the use of AI in these three areas have already been covered by others.
The last approach, single-cell foundation models, and in silico perturbation, is the AI Virtual Cell, or AIVC which is a topic a few of the presentations at Bio-IT World touched upon. It was something I was ready to learn more about at this conference, but was very much on the margins. (For those interested, here is a solid 2024 Perspective in the journal Cell that covers building a virtual cell with artificial intelligence with some 42 authors, all leaders within this single cell movement.)
Dr. Jenkins walked through an illustration of a party with a group of people in it. Who normally associates with whom? And then what if you had >100 million other observations of that party with the same people? Thus a Generative Pretrained Transformer (GPT) can Generate new sentences (“G”), Pre-train on massive data (“P”) and apply to new contexts via a Transformer (“T”). When used to analyze single cell transcriptome scRNA-Seq data, "genes are words, and cells are sentences".
He showed the large number of scRNA datasets from the scAtlas, with >100M cells (hearkening back to the >100M observations of the party in the prior slide), and enumerating the number of cells from tissues (33M from brain, 1.8M from heart etc). The scAtlas has no less than 39 organs represented. With this dataset, the GPT gets applied to all the collection of scRNA-Seq data from both healthy and diseased tissues.
The resulting output from the GPT is 1) accurate cell clustering, 2) elucidation of gene regulatory networks, and 3) healthy and disease cell predictions of perturbations, with genes knocked out or knocked in.
Here’s where the headline spoke volumes. “In silico perturbation (sometimes) corroborates in vitro data”. Sometimes! “You can see the trends, but directional and not absolute.” For one example, Geneformer (Theodoris et al.) shows in silico deletion of GATA4 and TBX5 perturbs validated co-bound target genes via ChIP-Seq. He also brought up scOTGM (Demir et al.) and scGPT (Cui et al.) He mentioned the “hot trend is to bash them” while at the same time he used the word “(sometimes)” in the title of the slide. “The jury is still out on whether these are working as advertised.”
The irony of the words “sometimes” and “bashing” was not lost on this attendee.
At this juncture he raises a question: why simulate perturbation data? He provides three answers: in silico perturbations (knock-down and knock-in) can be conducted genome-wide in all cells and cell types in tissues; they can be fine-tuned on disease versus healthy cells and cells from tissue, even post-mortem; and CRISPR screens combined with sequencing (i.e. Perturb-Seq, CROP-Seq) but are “great however expensive and limited to amenable cells”.
An important caution is applied here. Healthy scRNA-Seq data may not necessarily extend to other possible perturbation states. Secondly actual perturbation data is very limited (compared to scAtlas). Third, perturbation data generates gene lists. He enunciates the words “gene lists” like he was describing a boorish and annoying uncle you see every year at Thanksgiving.
Here he uses the gene list generation as a cue to refer to Causaly as Novartis’ LLM tool applied to the biomedical literature. Over 3,300 researchers within their company use Causaly to explore 400M biological relationships. He showed a query for malaria, with the targets “could be essential at different life cycle stages of the parasite”, then produced a 10 page “Target Assessment” report with 29 proposed target classes. It took less than two minutes, while it would have taken him “easily a week” by hand. No less than 32 documents were referenced, and about 60 “re-prompted searches” (where the LLM automatically ran additional queries).
Returning to the in silico perturbation model, downstream of the scRNA data are ‘genetics and genomics’ confirming expression in patients and models, then through Causaly LLM and OpenTargets supporting research, reducing the gene list down to “72 gene candidates” (as a target hypothesis).
To validate these candidates, an advanced organoid disease model is utilized however every gene cannot be tested in an organoid due to laboratory and budget limitations; compound screens and functional genomics screens progress from there. It is not more efficient. It does have more hypotheses than they otherwise would have had. He points out that “it always gets back to the lab” where the laboratory wet-work is the bottleneck.
He said there are “not many shortcuts to target validation. You solve one thing, the bottleneck shifts.” “Experimental model systems and target validation remain a bottleneck and limit the speed at which AI can accelerate targets into portfolios.”
Small molecule design
He began the second section with a slide titled “Drug hunting: how to identify the best candidate from 1060 potential molecules?” You generate ligands, you need a synthesize-able molecule, you need a binding pose (or co-fold), and then you predict binding affinity to know what to synthesize first. AI addresses all three.
He explained the principle in a slide called Generative Chemistry: start with a non-optimal seed, progress through encoding, a multi-objective optimization in latent space, then lastly decoding into an improved compound. The middle (“latent space”) is multidimensional and continuous, trained on millions of 3D structures.
He then showed a timeline of progress: the first 3D GenChem model in 2020 called LiGAN, where Novartis took a look at them and found in their benchmarks a “low drug likeness”. 2021 another model called 3D-SBDD. Jenkins took this opportunity to acknowledge out how important open-source algorithms are to commercial drug discovery. “Hats off to academics, this is how things get started.”
Then in 2022: Pocket2Mol, labeled in red: “much improved” in their benchmark, and explored by them (which in their iteration they call Pocket Crafter). Pocket2Mol is an autoregressive generative GNN model, using “frontier atoms” that sample across atoms in their locale, and exploring if the resultant molecule can fit into a pocket. 500K molecules tested to fit, likened to a Jenga puzzle or even Candy Crush; which pieces fit into a constrained space.
Applying it to a famous target the MYC proto-oncogene and its protein interaction partner WDR5, can a protein interaction be disrupted? Their historical work used 1.1 million compounds and came up with 2,715 hits. Pocket Crafter (Shen et al.) suggested 2,029 compounds, of which 7 were hits. This was a 13-fold improvement. Looking up the details in the reference, 3 of the compounds, labeled PC-1, -2 and -3, showed IC50 values of 27-36 uM.
They had knowledge of which residues were interacting with the molecule; AI found novel residues. AI found many co-folding methods. “It has been a game-changer” to have these generative models. A dynamic tool brings a new dimension to small-molecule binding prediction.
These affinity models are known as interaction methods. They represent proteins with secondary and tertiary features, in conjunction with the compounds. The joint models learn from the chemical features, and you can improve the affinity prediction; he suggested it may further be improved by adding physics to the AI approach.
Expanding on the co-folding approach, he presented a set of six open source generative co-folding models: AlphaFold3, RoseTTAFold All-Atom, Umol, Chai-1, OpenFold3, and Boltz-2. The value of these models being open is clear; however the business opportunities will reside downstream, similar to what happened in genomics with the sequencing becoming commoditized and the business opportunities moved to library prep, analytics and clinical interpretation. Concluding this section, Jenkins did point out two remaining challenges: ranking ligand designs by affinity prediction remains a challenge at scale, and synthesis of these de novo designs are a bottleneck in DMTA (design, make, test, analyze).
He made a footnote here of interest: these methods can make families of molecules, but the future will be more autonomous chemistry platforms with the ability to make single molecules.
Assay design
The third and final section started with the statement, “High content screening is a key technology for drug discovery”. Automated microscopy, also known as high content imagers, take snapshots of fixed cells stained with specific fluorescent antibodies, and these images have their features extracted. The drug discovery use cases are manifold: phenotypic screening, determination of mechanism of action, triage or prioritization of molecule hits, determine off-target effects or safety signals, and SAR, or structure/activity relationships, which is the relationship between chemical structure and biological activity.
Dr. Jenkins pointed out these methods fix cells in time, are labor intensive, and you have to move on to other cells to study because those samples have been consumed.
With in silico labeling, AI can be used to predict from brightfield images what the staining will be. Analogous to automatic ‘colorizing’ of black and white photographs, there’s enough context in the photograph to determine hair color, skin color, and the color of objects in the background. The same concept is applied to cell images, and of course these cells can remain in their native growth media.
He used a Schwann cell myelin sheath (protective) illustration, how a drug can delay demyelination or help regrow the myelin. With an antibody assay, the antibody ‘crashes out’ (becomes insoluble) whereas they are able now to predict fluorescence staining from brightfield images. This techniques enables ‘kinetic imaging’, as live cells can be imaged and subsequently analyzed. He showed data where the prediction had an R2 of 0.830 to fixed and stained cell imaging.
The benefit of this method: The “kinetic reads” are non-destructive (sample sparing); reduction by 80% of reagent consumption, reduction of screening time by 10-fold; and scalable to multiplex screening in silico from brightfield images. These are remarkable efficiencies.
Beyond labeling, Novartis researchers took this a step further. Jenkins showed an AI-generated image of an astronaut riding a horse, where a tool that uses prompts to create images is called Stable Diffusion. Applying this concept to a high content image (he used an image of U2-OS cells treated with a Chk1 inhibitor), they developed a diffusion model conditioned on compound bioactivity profiles for predicting high-content images, a method they call pDIFF. They used predicted QSAR (quantitative structure activity relationship) values for >10,000 dose-response assays, developed an in silico bioactivity profile, developed a diffusion model to go from profile-to-image.
{kind=link}
And instead of a novel image, one of an astronaut riding a horse, the pDIFF tool creates a stained cell image.
Using a set of 3,750 MoA (mechanism of action) compounds profiled in the U2-OS cell painting assay, he laid out a 90% training / 10% test set protocol, and showed data how similar the pDIFF generated images were compared to the ‘real’ ones (traditionally fixed and stained). Remarkable work!
And then they could run the diffusion model in reverse; retrieve molecules inducing similar phenotypic outcomes back to known active molecules. (This part I’m going to need to think about some more.) Their first project had measurable impact with this method in early 2025, with a 12-fold hit enrichment and “additional hit expansion ongoing”.
He summarized this section by pointing out a new bottleneck: do we need new models for every cell type and stain used?
What will AI do for Drug Discovery?
Three sections, three bottlenecks identified.
He concluded this plenary presentation with a graph, with creativity on the X-axis and Efficiency on the Y. Traditional Drug Discovery was closer to the origin. “AI Ambition” is up and to the right. He placed “Combinatorial Libraries?” to the left of Traditional Drug Discovery (less creative) but above on the Y-axis (more efficient).
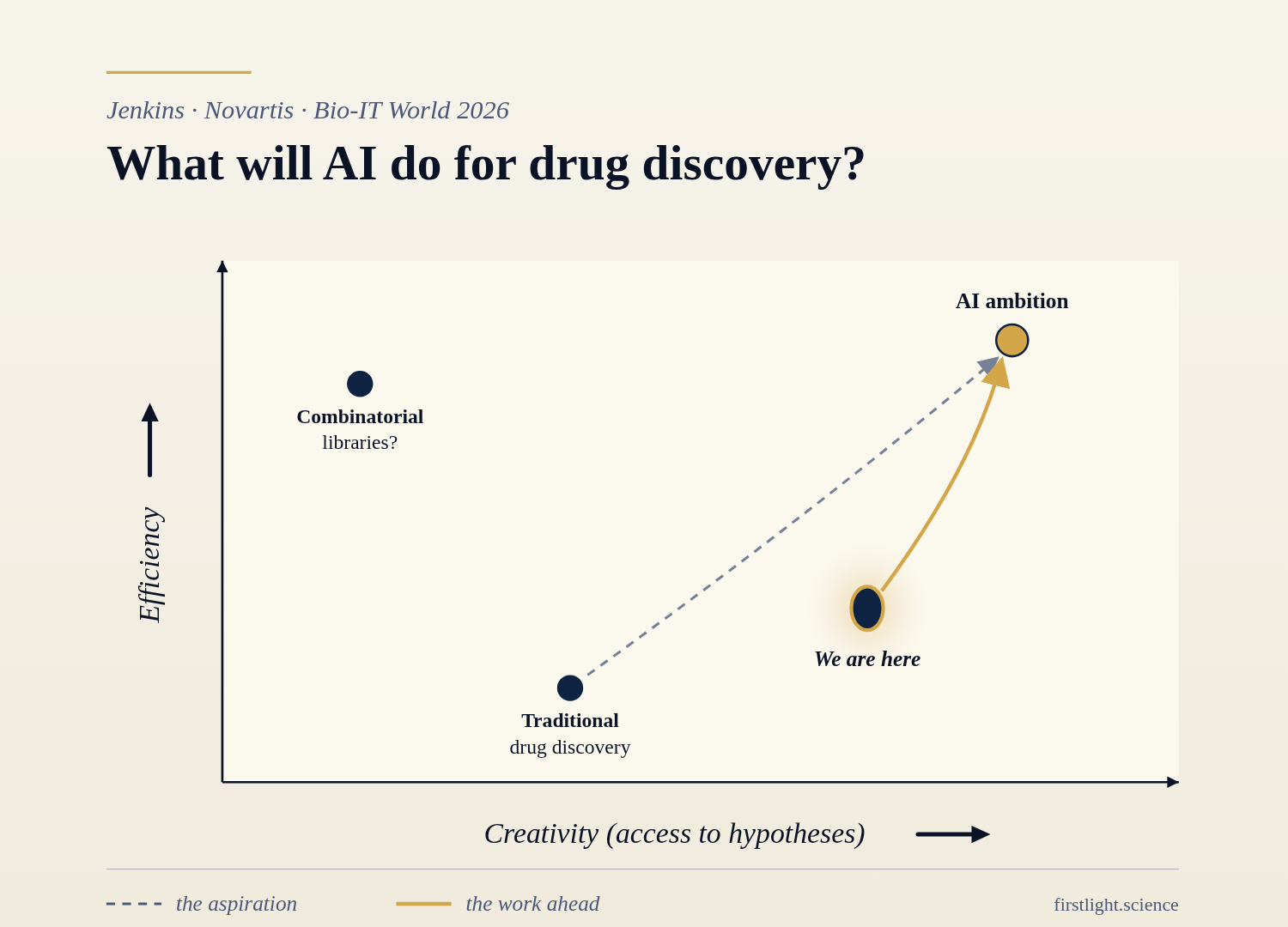
“We are here” is to the right (increased creativity) and slightly above on the Y-axis compared to Traditional Drug Discovery.
Ambition’s goal for AI is to test fewer targets, and make fewer molecules. How? Method maturity, successes and trust, and adoption.